
— Gemeinsame Studie von Zhejiang CDC, Macro & Micro-Test und China CDC, veröffentlicht in Frontiers in Cellular and Infection Microbiology
Studienübersicht
Im Mai 2026 veröffentlichte Frontiers in Cellular and Infection Microbiology (JCR Q1, IF ≈ 4,6) eine Studie unter der Leitung des Zhejiang Provincial Center for Disease Control and Prevention (Zhejiang CDC) mit Beteiligung des Bioinformatik-Teams von Beijing Macro & Micro-Test Bio-Tech Co., Ltd. und des National Institute for Communicable Disease Control and Prevention (China CDC). Der Titel der Studie lautet:
„Identifizierung und phylogenetische Analyse von sieben Brucella abortus-Stämmen in Zhejiang, China.“

Diese Studie stellt die erste systematische, auf dem gesamten Genom basierende phylogenetische Rückverfolgbarkeitsanalyse von Brucella abortus (B. abortus) in der chinesischen Provinz Zhejiang dar. Das Forschungsteam analysierte sieben Isolate, die zwischen 2015 und 2025 gesammelt wurden (vier Stämme humanen und drei bovinen Ursprungs aus Jinhua, Quzhou und Ningbo). Die Ergebnisse liefern genomische Belege für den Ursprung und die Übertragungswege dieser im Norden dominanten Spezies in einer für südliche Epidemiegebiete ungewöhnlichen Region Ostchinas.
Hintergrund und Bedeutung
Brucellose ist eine Zoonose, die durch Bakterien der Gattung Brucella verursacht wird. Brucella abortus infiziert vorwiegend Rinder, kann aber auch Menschen erkranken lassen. In China zeigt die Brucellose deutliche geografische Unterschiede: Die höchste Inzidenz tritt in den nördlichen Provinzen (z. B. Innere Mongolei, Shanxi, Heilongjiang) auf. Im Gegensatz dazu dominieren in den südlichen Provinzen, darunter Zhejiang, seit jeher Brucella melitensis, während Fälle von B. abortus nur sehr selten gemeldet wurden. Diese regionale Diskrepanz macht die genetische Charakterisierung und die Herkunftsermittlung von B. abortus in Zhejiang zu einer zentralen Priorität für die öffentliche Gesundheit.
Methoden und wichtigste Ergebnisse
Das Forschungsteam verfolgte eine vielschichtige Strategie, die Molekularbiologie und Bioinformatik kombinierte:
1.Pathogenidentifizierung und grundlegende Typisierung
Die PCR-Analyse des BCSP-31-Gens und die AMOS-PCR bestätigten, dass es sich bei allen sieben Isolaten um B. abortus handelte.
Die Multilocus-Sequenztypisierung (MLST) auf Basis von neun Haushaltsgenen ergab, dass alle Isolate zum Sequenztyp ST2 gehörten, was auf eine hohe genetische Homogenität der zirkulierenden B. abortus-Stämme in Zhejiang hinweist.
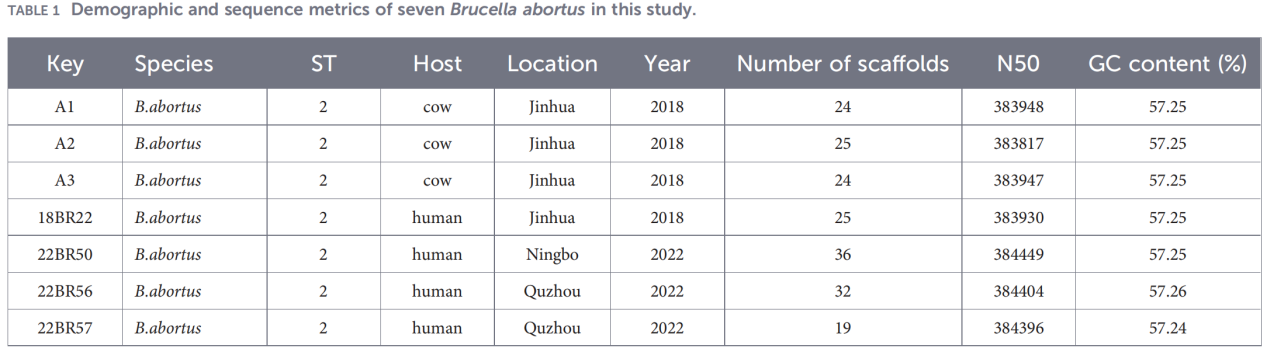
2.Charakterisierung des gesamten Genoms
Die Sequenzierung des gesamten Genoms erfolgte auf der Illumina NovaSeq-Plattform. Die Analyse der durchschnittlichen Nukleotididentität (ANI) zeigte, dass die Zhejiang-Isolate eine bis zu 99,99%ige Ähnlichkeit mit dem Referenzstamm B. abortus 544 aufwiesen.
Die Pan-Genomanalyse ergab eine hochkonservierte Population: Es wurden 3.084 Kerngene identifiziert, dazu nur 10 Hüllengene, und es wurden keine Weichkern- oder Wolkengene nachgewiesen.
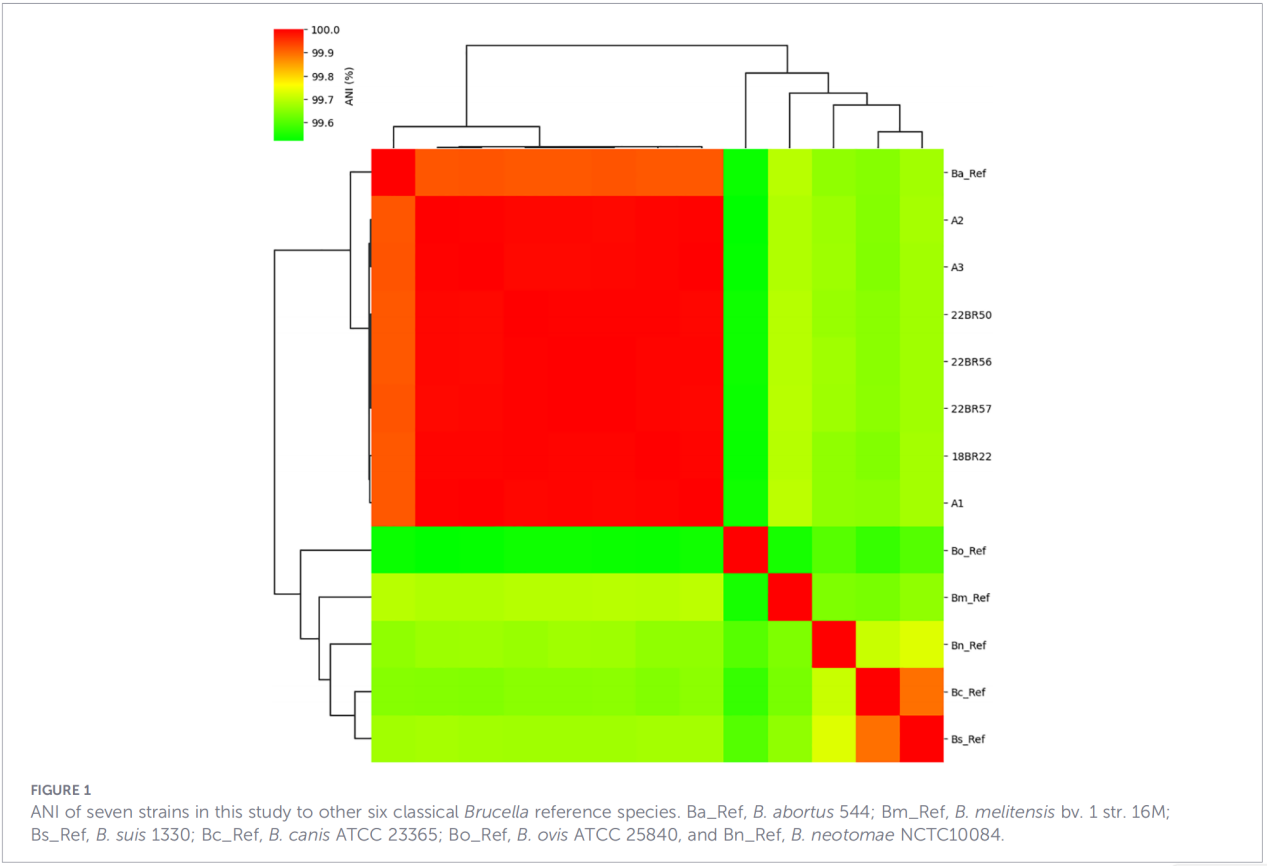
3.Virulenz- und Antibiotikaresistenzgenprofile
Insgesamt wurden 68 Virulenzfaktoren vorhergesagt, die klassische Signalwege wie die LPS-Biosynthese, das T4SS-Sekretionssystem und das Zwei-Komponenten-Regulationssystem BvrR-BvrS abdecken. Bemerkenswerterweise fehlten allen Isolaten die Adhäsingene bmaA und btaF. Die Resistenzgenanalyse detektierte in der CARD-Datenbank lediglich das mprF-Gen; weitere Resistenzdeterminanten wurden nicht identifiziert.
 4. Phylogenetische Rekonstruktion und Vererbungsverfolgung
4. Phylogenetische Rekonstruktion und Vererbungsverfolgung
Die Analyse von Einzelnukleotid-Polymorphismen (cgSNP) des Kerngenoms ordnete die Zhejiang-Isolate einer spezifischen Position im globalen phylogenetischen Baum zu. Die Ergebnisse zeigten, dass die Zhejiang-Stämme zusammen mit Stämmen aus Russland, der Mongolei und mehreren nordchinesischen Provinzen (Ningxia, Heilongjiang, Innere Mongolei, Hebei, Gansu, Peking) eine monophyletische Gruppe bilden. Diese Gruppe spaltet sich weiter in drei distinkte Subkladen (Klade 1–3) auf, was auf mehrere unabhängige Einschleppungsereignisse hindeutet.
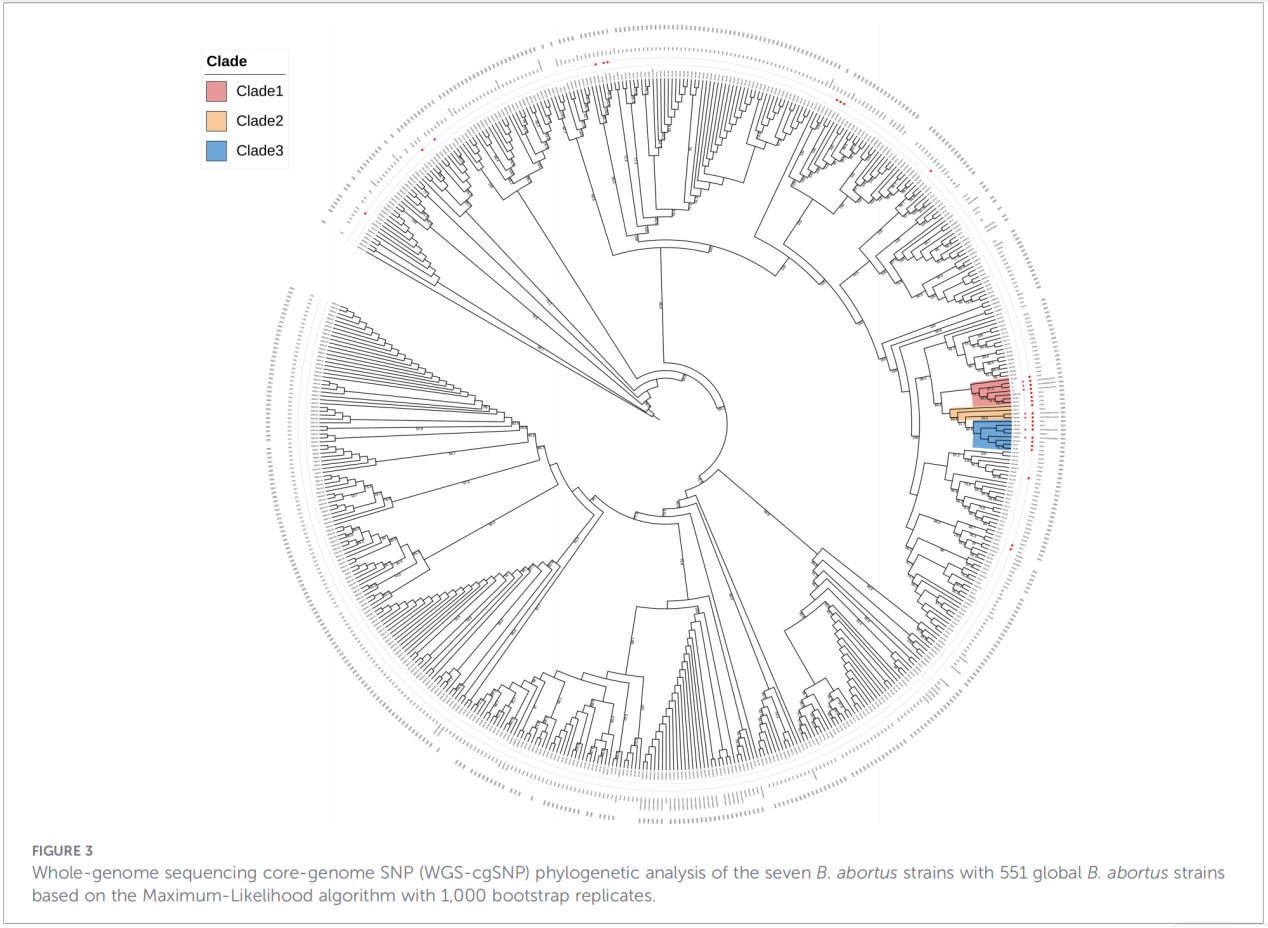
Schlussfolgerungen und Implikationen
Diese Studie liefert den ersten hochpräzisen Genomdatensatz von B. abortus in der Provinz Zhejiang und führt zu mehreren wichtigen Schlussfolgerungen:
- Clear genetischer Hintergrund– Die in Zhejiang zirkulierenden B. abortus-Stämme gehören zum ST2, sind genomisch hochkonserviert und stellen eine typische bovine Brucellose-Linie dar.
2. EviHäufigkeit der regionenübergreifenden ÜbertragungDie phylogenetische Analyse stützt nicht die Existenz einer eigenständigen endemischen Linie in Zhejiang. Die Daten legen vielmehr nahe, dass diese Stämme aus Nordchina stammen und möglicherweise einen gemeinsamen evolutionären Hintergrund mit Stämmen aus Russland und der Mongolei teilen. Das Vorhandensein von drei Subkladen deutet auf mehrere voneinander unabhängige Einschleppungsereignisse hin.
3. Auswirkungen auf die öffentliche GesundheitDie Ergebnisse unterstreichen den Wert der genomischen Überwachung von Brucellose selbst in traditionell nicht endemischen Regionen wie Zhejiang. Obwohl die Fallzahlen derzeit niedrig sind, können hochauflösende Methoden wie cgSNP die Quelle importierter Ausbrüche effektiv zurückverfolgen und wissenschaftliche Belege liefern, um Übertragungsketten im Zusammenhang mit dem Transport von Nutztieren zwischen den Provinzen zu unterbrechen.
Diese Arbeit schließt nicht nur eine Forschungslücke in der Provinz Zhejiang, sondern liefert auch neue Basisdaten für die Erregerüberwachung und Risikobewertung der Brucellose in der Jangtse-Delta-Region.
Papierinformationen:
Yang, Y., Shi, X., Chen, J., Wang, L., Wu, Z., Yao, W., … & Wu, B. (2026). Identifizierung und phylogenetische Analyse von sieben Brucella-abortus-Stämmen in Zhejiang, China. Frontiers in Cellular and Infection Microbiology, 16, 1758965.
Veröffentlichungsdatum: 10. Juni 2026